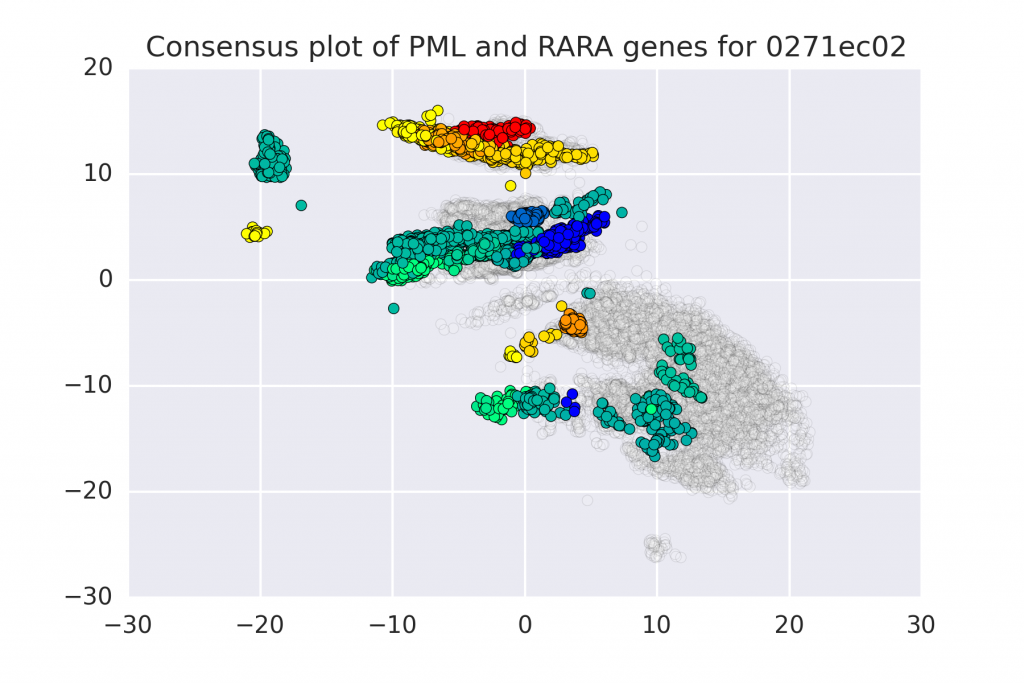
In this work we propose a method to compute continuous embeddings for kmers from raw RNA-seq data of the transcriptome, without the need for alignment to a reference genome. The approach uses an RNN to transforms kmers of the RNA-seq reads into a 2 dimensional representation that is used to predict abundance of each kmer. In this latent transcriptome we observe the grouping of kmers that corresponds to the genes that they are expect to belong to. More info:
Paper Slides
Assya Trofimov, Francis Dutil, Claude Perreault, Sebastien Lemieux, Yoshua Bengio, Joseph Paul Cohen. Towards the Latent Transcriptome. 2018, http://arxiv.org/abs/1810.03442.